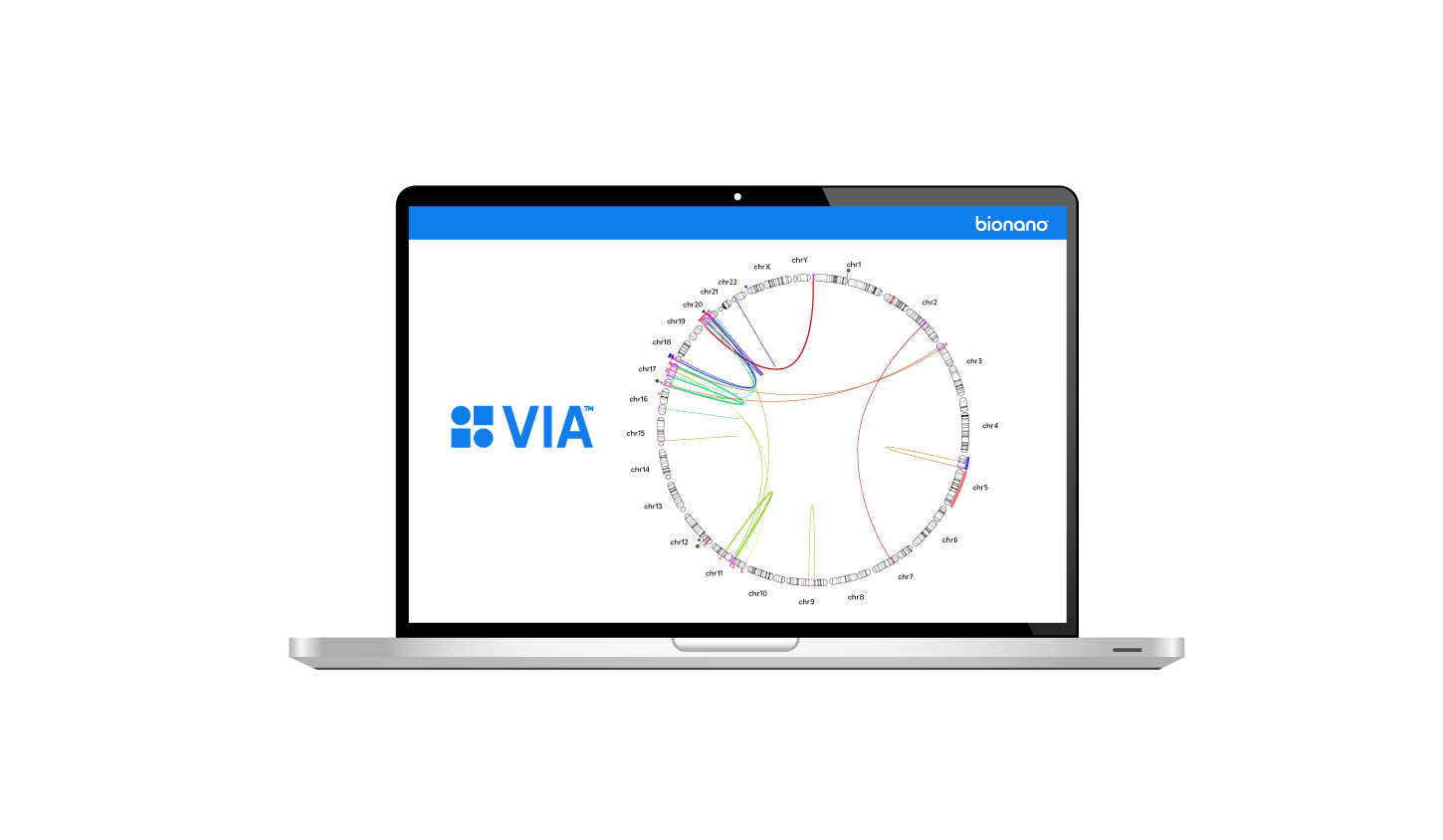
Watch and listen as the scientists in the Bionano community share their latest research findings and detail their experiences with our solutions.
Show More Results