Find Patterns and Uncover Answers to Important Biological Questions
The Nexus Copy Number software enables researchers to analyze multiple data types—arrays, NGS panels, WES/WGS, and expression data—across research cohorts for a genome-wide view. Reveal new insights with incredible simplicity through a variety of advanced analysis and visualization tools.
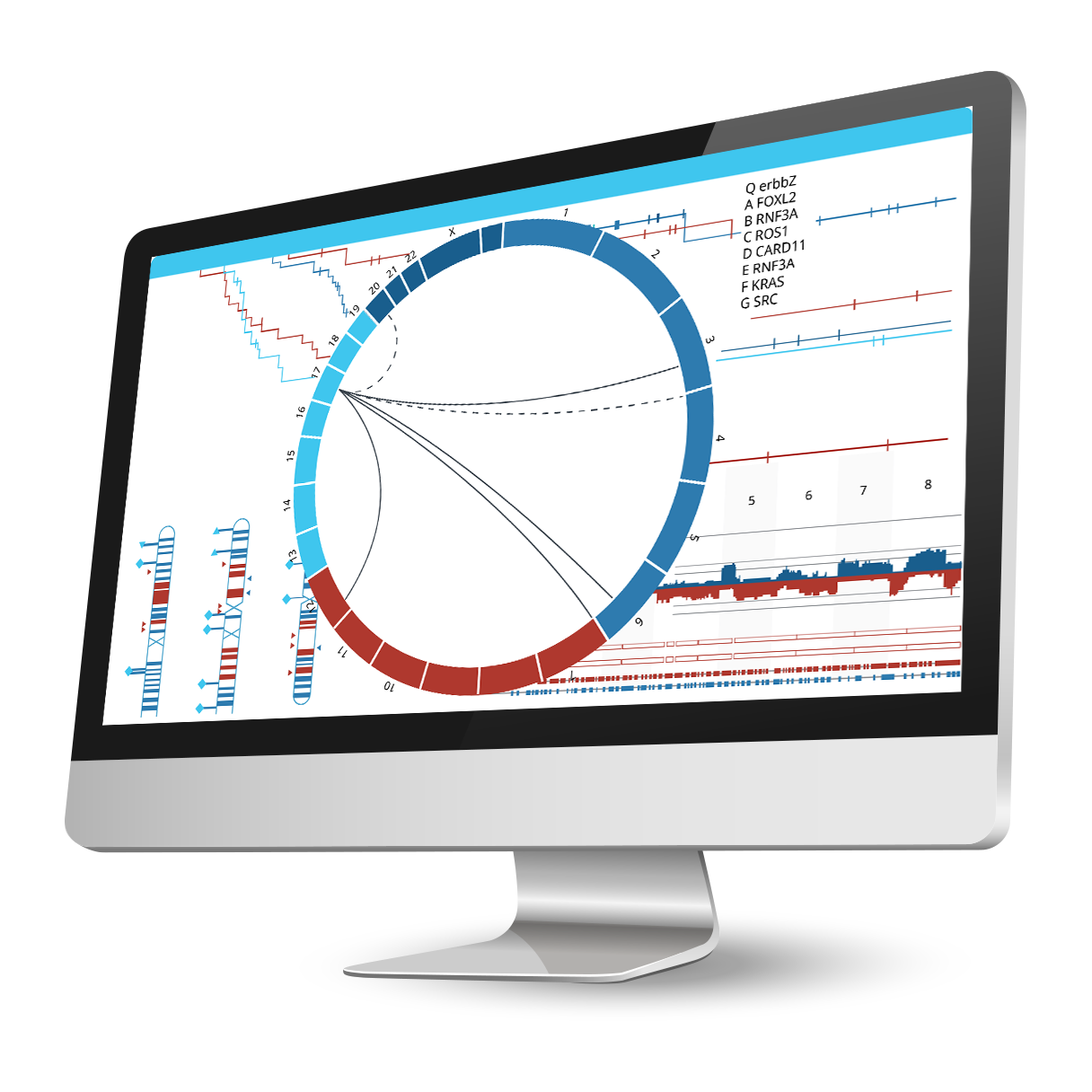
Integrate phenotypic and genomic data to uncover significant genetic correlations.
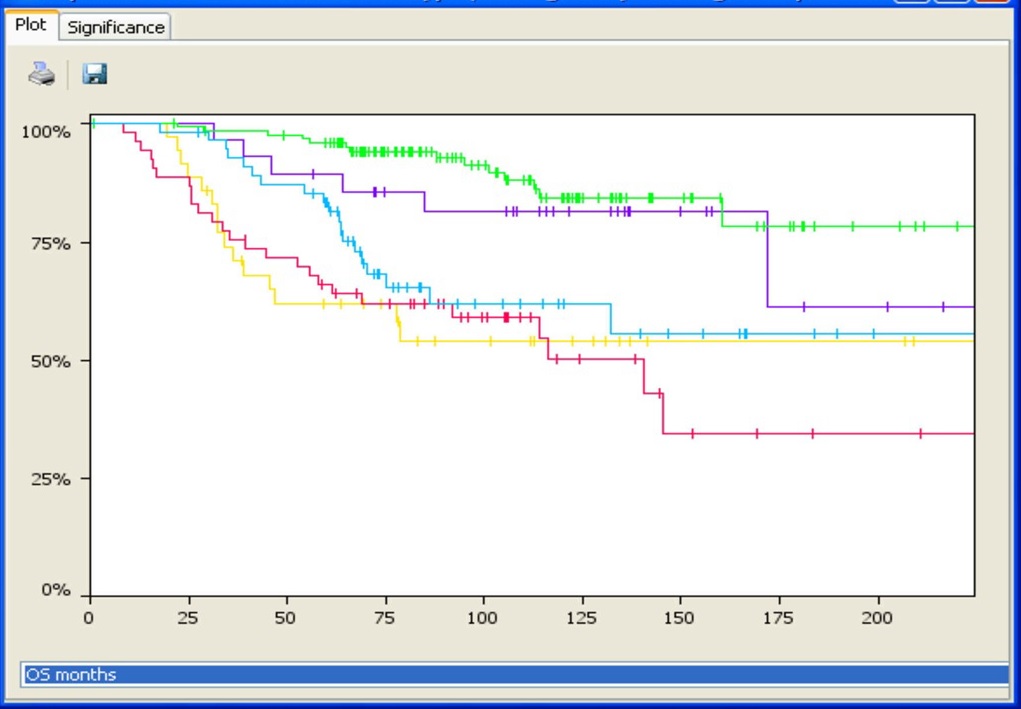
Associate an unlimited number of sample annotations or attributes for further downstream analyses. Use the predictive power tool to find correlations between phenotypic attributes and genomic changes, perform survival analysis for cancer samples (with Kaplan-Meir plots), or stratify samples based on aberration profiles and phenotypes to identify driver mutations. Nexus Copy Number enables grouping using K-means or hierarchical clustering with the ability to adjust the number of clusters. Identify gene ontology (GO) terms enriched over the entire genome using GSEA or find GO terms significantly over-represented in selected regions only. Use the concordance function to identify genomic alterations co-occurring with copy number changes, LOH regions, or small mutations, and depict these relationships in a circle plot.
Analyze and visualize multiple samples in parallel—no bioinformatics expertise required.
Nexus Copy Number provides you with robust statistical functions and rich interactive graphics, without the need for graphical and statistical programming expertise. It is a powerful desktop (Windows or Mac OS) solution allowing visualization and analysis of tens of thousands of high-density arrays in parallel. Many statistical functions are built in, allowing you to easily perform large cohort analyses as well as individual sample analysis and visualization. Analysis settings allow flexible and customized analysis.

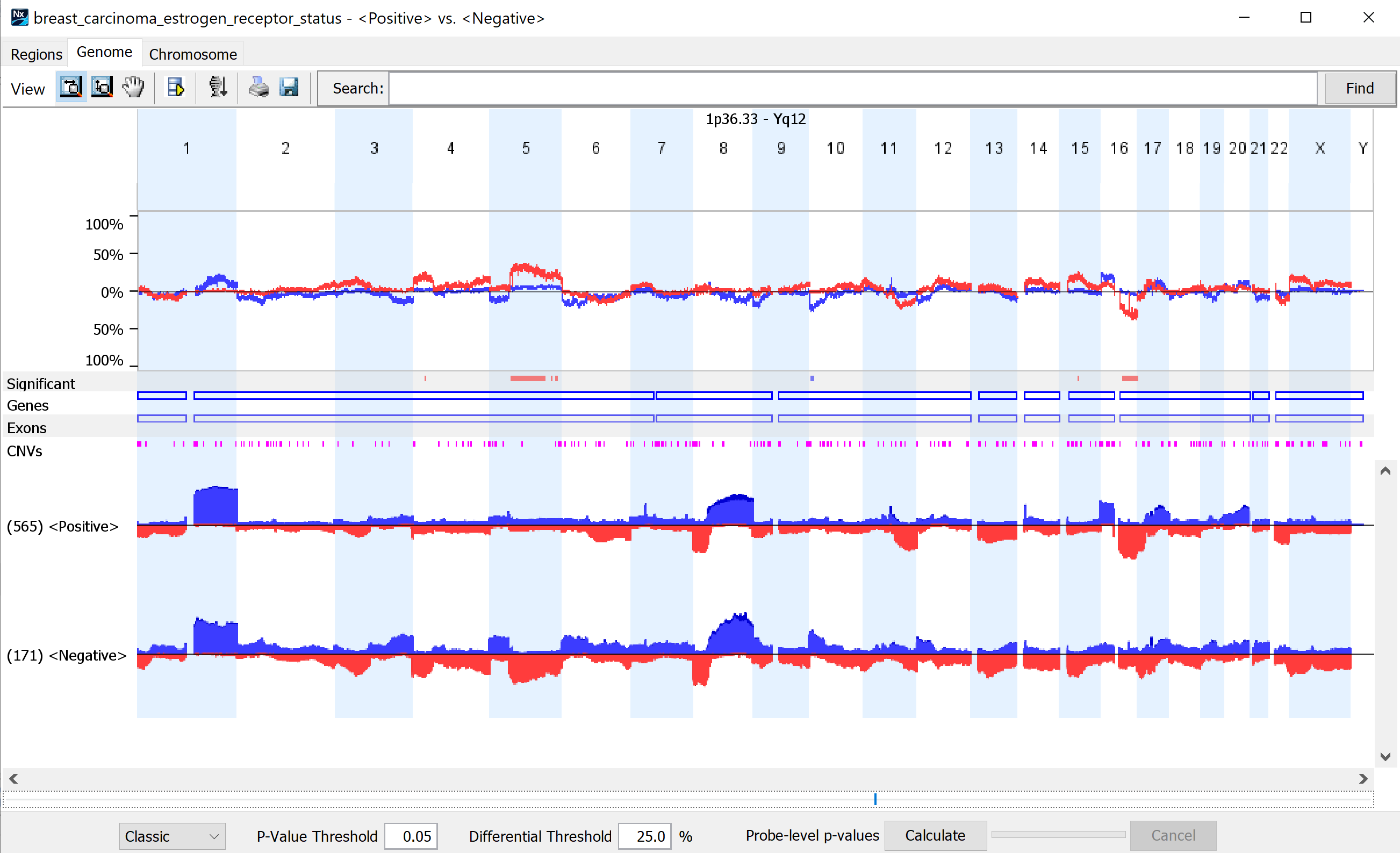
Compare genomic profiles between subgroups to identify statistically significant differences.
Find statistically significant genomic changes between biologically important subgroups using one of several different comparison types available (i.e., paired, sequential). Results can be viewed in table format or interactive graphical displays. View frequency plots of designated subgroups in parallel, along with indicators displaying those regions that are significantly different.
Identify important regions of genomic variation by integrating mRNA, miRNA, sequence variants, methylation, and copy number changes.
Gene expression results can be loaded into the software, allowing more complete analysis by integrating multiple modalities. Sequence variants from MAF or VCF files allow the analysis of point mutations and indels, either alone or integrated with copy number changes. Results from expression arrays or RNA-seq—along with significance and fold change—can be integrated on a per-sample basis to identify genes with coordinating changes in gene expression and copy number or allelic events.


Query for region/gene across multiple projects from GEO and TCGA via Nexus DB repository.
Nexus Copy Number software is bundled with the Nexus DB genomic data repository, allowing querying of similar aberrations across thousands of samples from GEO, TCGA, and more. Search for keywords to find similar projects or samples with specific annotations. You can share data and collaborate with colleagues using a secure data-sharing scheme. Add the TCGA Premier product, allowing access to high-quality and manually curated data from TCGA for deeper studies involving survival analysis or tumor sub-type profiling.
Related Products




